
16x24x4 Air Filter | MERV 8 - 16x 24x4 6 pack
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you've safely connected to the .gov website. Share sensitive information only on official, secure websites.
Articles from The Journal of Biological Chemistry are provided here courtesy of American Society for Biochemistry and Molecular Biology
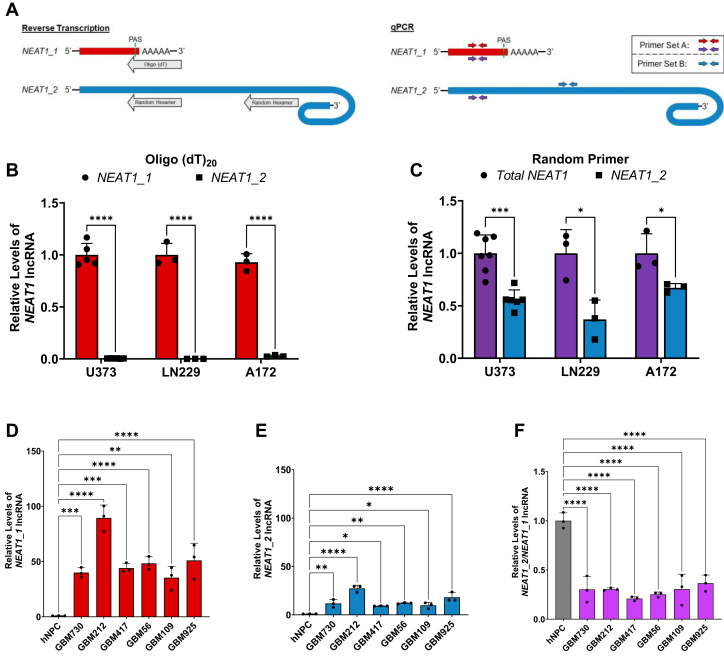
A common problem in understanding NEAT1 dysregulation is a lack of means for the distinct quantification of NEAT1_1 by the commonly used quantitative RT-PCR (RT-qPCR) approach, because NEAT1_1 completely overlaps with the 5′ end of NEAT1_2. Numerous studies reported dysregulation of NEAT1_1 based on RT using random primers, followed by quantitative PCR (qPCR) which amplifies the 5′ end sequence shared by both NEAT1_1 and NEAT1_2, hence measuring Total NEAT1 instead (28, 32). Such an experimental caveat results in misleading and sometimes controversial conclusions. To address this problem, we employed an assay that takes advantage of the different 3′ ends of the NEAT1 isoforms to distinctly quantify NEAT1_1 and NEAT1_2 at the steady state levels in human GBM cell lines. As shown in Figure 1A (left), NEAT1_1 is cleaved at a conventional PAS and polyadenylated, while NEAT1_2 contains a structured 3′ end that does not include a poly(A) tail.
An amount of 1 μg of total RNA from three biological replicates of U373 parent and NEAT1 ΔPAS clones (1 and 2) was used for poly-A-enriched RNA-seq library preparation using the TruSeq Stranded mRNA kit (Illumina, 20020594). Libraries were sequenced on an Illumina HiSeq platform (Admera Health, LLC) with a read length configuration of 150 paired end, targeting 80 M total reads per sample.
The high-throughput sequencing data, including RNA-seq, have are available at the Gene Expression Omnibus under the accession number GSE262598.
DEGs with significantly changed splicing efficiency were identified using unpaired Student’s t test with p < 0.05 as a cut-off.
ASOs engineered to target and KD NEAT1_2 were created and purchased from integrated DNA technologies. The ASOs were phosphorothioate modified at the backbone and the five terminal nucleotides on the 5′ and 3′ ends were substituted with 2′-O-methoxyethyl ribonucleotides. The sequences of the NEAT1_2 and negative control ASOs used are shown in Table S3. The ASO-targeting NEAT1_2 or the negative control were transfected using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) into U373 control and NEAT1 ΔPAS cells and harvested after 24 h for RNA analysis. A final ASO concentration of 200 nM was used for all transfection reactions.
NEAT1_2 is well established as an architectural lncRNA, necessary for the formation of nuclear paraspeckles in a subclass of mammalian cells (32, 37, 38, 59). Various regions of NEAT1_2 bind and organize RBPs known as paraspeckle proteins (27, 28, 29, 32, 35, 36, 43, 44). Previous studies reported that transient KD or elevated NEAT1_2 achieved by blocking the proximal NEAT1 PAS with ASOs can alter the number and/or size of paraspeckles (29, 58, 71, 77). Hence, paraspeckles must be dynamic, in response to changes in NEAT1_2. While paraspeckle formation has been studied in a wide range of cell types (32, 35, 58, 77), our studies provide the first evidence that paraspeckles form in GBM cells. Moreover, the long-term increase in NEAT1_2 levels, as a result of the permanent loss of the NEAT1 PAS, led to the sustained increase in paraspeckle numbers in GBM cells, without affecting paraspeckle size (Fig. 3). Previous studies have shown NEAT1_1 is present in, but not essential for paraspeckle formation (27, 28, 44). However, whether deficiency of NEAT1_1 may affect the composition and function of paraspeckles, which are indicated in cellular stress responses, cell differentiation, and cancer progression, is not known (40, 41, 42). Interestingly, regardless of the essential function of paraspeckles in RNA processing (38, 78), many RNA processing pathways were downregulated in the NEAT1 ΔPAS GBM cell line which harbored elevated NEAT1_2 along with diminished NEAT1_1 (Fig. 6). These results raised a question as to whether NEAT1_1 is also important in governing paraspeckle integrity and function, which warrants further investigation in future studies.
Paired end RNA-seq reads were mapped to human genome assembly version (GRCh38/h38) using TopHat version 2.1.0 with default parameters (89). Aligned reads within the bam file were sorted based on genomic coordinate by SAMtools (90). Differential gene analysis was executed using Cuffdiff version 2.1.1 (91). DEGs with FDR <0.05 were indicated as significant. GO enrichment analysis was performed by PANTHER online (https://www.pantherdb.org/) (92, 93, 94). Bubble plot display of GO terms with enrichment was generated with SRplot (95).
Previous studies have examined the abundance and function of NEAT1 in an array of cancers. These studies report that aberrantly increased NEAT1 is associated with tumor progression and poor prognosis (45, 46, 47). In non–small cell lung cancer, high levels of Total NEAT1 were found in non–small cell lung cancer tissue compared to control (48). Moreover, knockdown (KD) of NEAT1_2 was correlated with decreased cell proliferation and invasion (48). In hepatocellular carcinoma, KD of NEAT1 led to decreased cell viability and increased apoptosis (49). On the contrary, NEAT1 was repressed in de novo acute promyelocytic leukemia compared with healthy donors and NEAT1 KD blocked myeloid differentiation (50). These and numerous other studies have revealed the complex roles of NEAT1 in cancers.
Although extensive studies have explored the oncogenic roles of NEAT1, most studies focus on the function of NEAT1 in sponging specific miRNAs in the cytoplasm (33, 79). This is unlikely the function of NEAT1_2, which is restricted in the nuclei. Surprisingly, very few studies have addressed the impacts of NEAT1 on the human transcriptome. Only a limited number of studies have conducted RNA-seq and transcriptomic analysis upon manipulation of mouse Neat1, with fewer analyzing the impact of human NEAT1 (80, 81, 82, 83). To our knowledge, our studies provide the first characterization of GBM transcriptomic changes as a result of reciprocal alterations of NEAT1 isoform levels.
The WT and mutant reporter constructs were transfected into the U373 GBM cell line in parallel cultures. Expression of the reporters was visualized by the EGFP fluorescence 48 h after transfection. DNase-treated poly(A) RNA was extracted followed by RT-qPCR using two pairs of primers either flanking or downstream of the NEAT1 PAS (Fig. 5B). Due to the absence of endogenous NEAT1_2 in the isolated poly(A) RNA pool (Fig. 1), these primers specifically detect reporter transcripts which employ the SV40 PAS but are not cleaved at the NEAT1 PAS. Moreover, primers specific for the EGFP region (Fig. 5B) were used in parallel RT-qPCR reactions to detect total reporter transcripts, regardless of which PAS is used, as an internal reference.
More than 98% of the human genome consists of noncoding sequences, which are extensively transcribed to produce noncoding RNAs (ncRNAs) (1, 2). Long noncoding RNAs (lncRNAs) are a family of ncRNAs that harbor greater than 200 nucleotides (nt) but lack protein-coding abilities (3, 4). Like mRNAs, lncRNAs are transcribed by RNA polymerase II, often undergo 5′ capping, splicing, and 3′ end polyadenylation (5, 6). lncRNAs are expressed in various cell types and tissues, which regulate broad gene networks through diverse mechanisms at the transcriptional and post-transcriptional levels (7, 8, 9, 10, 11). Functionally, lncRNAs influence essential biological processes including cell-cycle regulation, cell development, migration, and apoptosis (12, 13, 14). Interestingly, lncRNAs are much less conserved across evolution compared to protein coding genes (1, 15). Moreover, many lncRNAs are preferentially expressed in the brain and dysregulated in numerous neurodegenerative diseases (16, 17, 18, 19, 20, 21), as well as glioblastoma multiforme (GBM), the most common primary brain malignancy (22, 23, 24, 25).
The authors thank Dr Archa H. Fox for the feedback and guidance on the RNA-FISH work and for sharing the NONO antibody. We thank Luke A. Knudson for the guidance with our Imaris analysis. We thank Dr Anita H. Corbett for the critical review of the manuscript and intellectual contribution. We also thank Dr M. Lee Cato for the critical review and feedback of the manuscript.
The biogenesis of the overlapping but distinct NEAT1_1 and NEAT1_2 isoforms is well defined (29, 32, 44, 59, 71). Moreover, dysregulation and the oncogenic function of NEAT1 have been extensively studied in various types of cancer including glioma (22, 23, 24, 53, 72). Although the most definitive experiment for distinguishing NEAT1 isoforms is Northern blot (28, 71), this classical method requires large amounts of RNA which is difficult to obtain from patient specimens. Thus, most studies used the sensitive and quantitative method RT-qPCR, with a 5′ primer set within NEAT1_1 and a 3′ primer set specific for NEAT1_2. Although specific quantification of each NEAT1 isoform was claimed (29, 71), upon careful examination, one would find that most studies did not measure the levels of NEAT1_1 but rather measured Total NEAT1 transcripts (28, 32, 53, 54, 55). The problem arises from the use of random primers in reverse transcribing total RNA, which produces cDNA from both NEAT1 isoforms. Hence, the PCR primers designed to detect NEAT1_1 will also detect NEAT1_2 due to the complete overlap of NEAT1_1 with the 5′ sequence of NEAT1_2. Moreover, many studies applied siRNA/shRNA that target the common region of NEAT1 but claimed specific KD of NEAT1_1. These misleading conclusions contribute to some conflicting reports and create a complicated picture of NEAT1 isoform dysregulation and function under various disease conditions.
To generate the NEAT1 PAS cleavage reporter, a region of the NEAT1 transcript spanning nucleotides 3235 to 4175 was amplified from BE(2)-M17 neuroblastoma cells via PCR followed by TA cloning using TOPO TA Cloning Kit. The plasmid was propagated in TOP10 Escherichia coli (Thermo Fisher, K450001SC) and the PCR insert was subcloned into a pEGFP-C2 vector using KpnI (Thermo Fisher Scientific, ER0521) and ApaI (Thermo Fisher Scientific, ER1411) restriction enzyme sites. The expected sequence in the reporter gene was confirmed by DNA sequencing of the plasmid. The predicted QREs upstream of the NEAT1 PAS were then mutated in the pEGFP-C2-NEAT1 cleavage reporter. Primers used for the first and second QRE mutations are provided in Table S3. Successful mutagenesis was confirmed by DNA sequencing of the plasmid.
All images were obtained using a Nikon Eclipse TE2000 (Nikon) widefield fluorescence microscope with a 60× objective. Z-series were acquired at 0.2 μm steps, and image stacks were deconvolved using AutoQuant X3 software (https://mediacy.com/autoquant-deconvolution/; Media Cybernetics) and a 3-D blind algorithm. All acquisition parameters were kept consistent for all samples. The “Coloc” module of the Imaris software (https://imaris.oxinst.com/; Bitplane) was used for analysis and quantification of paraspeckle number and paraspeckle area. All intensity thresholds were set the same across all samples. Representative images were prepared using the FIJI software package (https://imagej.net/software/fiji/; ImageJ), and dot plots were generated with GraphPad Prism 10.0 (https://www.graphpad.com/; GraphPad Software).
Loss of nuclear QKI-5 causes reciprocal changes of NEAT1 isoforms.A, schematic of the putative QKI recognition element (QRE) upstream of the human NEAT1 PAS. B, schematic depicting the deletion of QKI-5 exon 7c by CRISPR-Cas9. Scissors depict cleavage sites by sgRNAs. C, RT-qPCR detection of QKI-5 mRNA in the U373 ΔQKI-5 clone compared to control. Data are shown as mean ± SD from 6 biological replicates, normalized to β-actin, and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗∗p < 0.01. D, immunoblot analysis of QKI-5 protein levels in the U373 control, the two NEAT1 ΔPAS clones, and the ΔQKI-5 clone. eIF5 was used as a loading control. E–G, RT-qPCR detection of (E) NEAT1_1, (F) NEAT1_2, and (G) quantification of the ratio of NEAT1_2 to NEAT1_1 steady state in U373 ΔQKI-5 clone compared to control. Data are shown as mean ± SD from 3 or 4 biological replicates, normalized to β-Actin, and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗p < 0.05, ∗∗p < 0.01. NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site; QKI, quaking; RT-qPCR, quantitative RT-PCR; sgRNA, synthetic guide RNA.
Statistical analysis was conducted as described in the corresponding Figure legends. Comparisons between experimental groups were performed using the unpaired Student’s t test using GraphPad Prism 10.0 (GraphPad Software). Multiple t test comparisons were performed using the Student’s t test with Holm-Šídák multiple comparison’s test. Multiple-group comparisons were performed using one-way ANOVA with Dunnett multiple comparison’s test. R-studios was used to perform Pearson’s Chi-squared test. All data are presented as mean ± SD for at least three independent experiments, unless otherwise indicated. Statistical significance was indicated by ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
To elucidate how dysregulation of NEAT1 isoforms impacts the GBM transcriptome, we performed RNA-seq of the NEAT1 ΔPAS clones in parallel with the parental U373 control, which uncovered broad transcriptomic changes (Figs. 6A and S7A). As shown in Figures 6B, 3215 differentially expressed genes (DEGs) are upregulated and 3288 downregulated in the NEAT1 ΔPAS clone #1, while 3481 DEGs are upregulated and 3562 downregulated in the NEAT1 ΔPAS clone #2, respectively (false discovery rate [FDR] < 0.05, Fig. 6B). We identified 5038 common DEGs in both NEAT1 ΔPAS clones compared to parent control cells (FDR < 0.05, Fig. 6C and Table S2). Further analysis of DEGs in the NEAT1 ΔPAS clones revealed a strong correlation (R2 = 0.90, p < 2.2 × 10-16) (Fig. S7B), providing evidence that these two independent clones show significantly correlated transcriptomic changes. Among these common DEGs, 2448 transcripts were upregulated while 2590 transcripts were downregulated in both NEAT1 ΔPAS clones (Fig. 6C).
Importantly, loss of the QREs causes a significant increase of transcripts not cleaved at the NEAT1 PAS, which are detected by both primer pairs compared to the WT reporter (Fig. 5, C and D). This result clearly demonstrates that the QREs are critical for the efficient utilization of the NEAT1 PAS, which is essential for the biogenesis of NEAT1_1. These data suggest that mutating the QREs upstream of the NEAT1 PAS prevents QKI-5 binding, therefore ablating the effects of QKI-5 on modulating the usage of the NEAT1 PAS, which regulates NEAT1 isoforms.
The long noncoding RNA nuclear paraspeckle assembly transcript 1 (NEAT1) is involved in a variety of human cancers. Two overlapping NEAT1 isoforms, NEAT1_1 and NEAT1_2, are produced through mutually exclusive alternative 3′ end formation. Previous studies extensively investigated NEAT1 dysregulation in tumors, but often failed to achieve distinct quantification of the two NEAT1 isoforms. Moreover, molecular mechanisms governing the biogenesis of NEAT1 isoforms and the functional impacts of their dysregulation in tumorigenesis remain poorly understood. In this study, we employed an isoform-specific quantification assay and found differential dysregulation of NEAT1 isoforms in patient-derived glioblastoma multiforme cells. We further showed usage of the NEAT1 proximal polyadenylation site (PAS) is a critical mechanism that controls glioma NEAT1 isoform production. CRISPR-Cas9–mediated PAS deletion reduced NEAT1_1 and reciprocally increased NEAT1_2, which enhanced nuclear paraspeckle formation in human glioma cells. Moreover, the utilization of the NEAT1 PAS is facilitated by the RNA-binding protein quaking (QKI), which binds to the proximal QKI recognition elements. Functionally, we identified transcriptomic changes and altered biological pathways caused by NEAT1 isoform imbalance in glioma cells, including the pathway for the regulation of cell migration. Finally, we demonstrated the forced increase of NEAT1_2 upon NEAT1 PAS deletion is responsible for driving glioma cell migration and promoting the expression of genes implicated in the regulation of cell migration. Together, our studies uncovered a novel mechanism that regulates NEAT1 isoforms and their functional impacts on the glioma transcriptome, which affects pathological pathways of glioma, represented by migration.
RNA-FISH was conducted as previously described (96). Briefly, cells were grown on and fixed onto coverslips (Carolina, 633029) using 4% paraformaldehyde in 1× diethyl pyrocarbonate-phosphate buffered saline (DEPC-PBS) for 10 min at room temperature. Coverslips were washed with 1× DEPC-PBS at room temperature, followed by one wash with 2× saline sodium citrate (SSC) for 10 min at room temperature, and a final wash with prewarmed 2× SSC containing 10% formamide. Coverslips were incubated in prehybridization buffer for 1.5 h at 37 °C after which coverslips were incubated in hybridization buffer containing FISH probes (1:100) overnight at 37 °C. The next day, coverslips were washed with prewarmed 2× SSC containing 10% formamide, followed by additional washes with 2× SSC. Coverslips were incubated in blocking buffer for 1 h at room temperature. The coverslips were then incubated with a mouse mAb against NONO (35) at 1:500 dilution in blocking buffer for 1 h at room temperature. Coverslips were washed with 1× DEPC-PBS and then incubated with secondary anti-mouse Alexa Fluor 488 (Thermo Fisher Scientific, A11001) at 1:500 dilution and in blocking buffer for 1 h at room temperature. Coverslips were washed with 1× DEPC-PBS and mounted onto slides with ProLong Gold Antifade Mountant with 4′,6-diamidino-2-phenylindole (Life Technologies, P36935). The FISH probes used in this study include Human NEAT1 5′ Segment with Quasar 570 Dye (Stellaris, SMF-2036-1) and Human NEAT1 Middle Segment with Quasar 570 Dye (Stellaris, SMF-2037-1).
Reciprocal changes of NEAT1 isoforms broadly altered the GBM transcriptome.A, volcano plot of one NEAT1 ΔPAS clone indicates differentially expressed genes (DEGs) in U373 cells due to loss of the NEAT1 PAS. Blue dots represent DEGs with significant reductions, whereas red dots represent DEGs with a significant increase. Black dots represent genes that do not change significantly (FDR > 0.05) upon loss of the NEAT1 PAS. B, bar chart indicating the number of identified upregulated and downregulated DEGs in the two NEAT1 ΔPAS clones, respectively. C, Venn diagram shows significant overlap of increased and decreased DEGs between the two NEAT1 ΔPAS clones. D, bar plot indicating the proportion of DEGs with changed splicing efficiency in the two NEAT1 ΔPAS clones. E, Gene Ontology (GO) analysis of the common downregulated DEGs in the NEAT1 ΔPAS clones with FDR <0.05. F, GO analysis of the common upregulated DEGs in the NEAT1 ΔPAS clones with FDR <0.05. G and H, RT-qPCR analysis of representative (G) upregulated and (H) downregulated DEGs enriched in multiple GO terms. Data are shown as mean ± SD from 3 biological replicates, normalized to RPL13A, and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗p < 0.05, ∗∗p < 0.01. FDR, false discovery rate; GBM, glioblastoma multiforme; NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site; RPL13A, ribosomal protein L13A; RT-qPCR, quantitative RT-PCR.
Establishment of NEAT1 isoform specific qPCR detection assay.A, schematic of the NEAT1_1 and NEAT1_2 transcripts depicting two reverse transcriptions by oligo(dT)20 or random primers (left schematic) to distinctly analyze steady state isoform levels by RT-qPCR (right schematic). Red arrows indicate primer set A used to detect NEAT1_1 after RT with oligo(dT)20. Purple arrows are the same primers used to detect Total NEAT1 after RT with random primers. Blue arrows represent primer set B used for detection of NEAT1_2 specifically. B, reverse transcription using oligo(dT)20 primer followed by qPCR shows specific detection of NEAT1_1 and lack of NEAT1_2 detection in U373 human GBM cells, as well as LN229 and A172 GBM cell lines. Data are shown as mean ± SD from 5 (NEAT1_1) or 6 (NEAT1_2) biological replicates in the U373 cell line and 3 biological replicates from LN229 and A172 cell lines. Data are normalized to RPL13A and compared using the ΔΔCT method. Unpaired Student’s t test with Holm-Šídák multiple comparison’s test was used, ∗∗∗∗p < 0.0001. C, reverse transcription using random primers followed by qPCR shows detection of both Total NEAT1 and NEAT1_2 at steady state levels in U373, LN229, and A172 GBM cells. Data are shown as mean ± SD from 7 (NEAT1Total) or 6 (NEAT1_2) biological replicates in the U373 cell line and 3 biological replicates from the LN229 and A172 cell lines. Data are normalized to RPL13A and compared using the ΔΔCT method. Unpaired Student’s t test with Holm-Šídák multiple comparison’s test was used, ∗∗∗p < 0.001. D and E, detection of (D) NEAT1_1 by oligo(dT)20 RT-qPCR and (E) NEAT1_2 by random primer RT-qPCR in patient-derived human GBM GSCs. Data are shown as mean ± SD of 3 technical replicates from samples derived from six patients and one hNPC control. Data are normalized to RPL13A and compared using the ΔΔCT method. One-way ANOVA with Dunnett multiple comparison’s test was used, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. F, quantification of the ratio of NEAT1_2 to NEAT1_1 in six patient-derived human GBM GSCs compared to one hNPC control. Data are shown as mean ± SD of 3 technical replicates, normalized to RPL13A, and compared using the ΔΔCT method. One-way ANOVA with Dunnett multiple comparison’s test was used, ∗∗∗∗p < 0.0001. GBM, glioblastoma multiforme; GSC, gliomasphere culture; hNPC, human neural progenitor cell; NEAT1, nuclear paraspeckle assembly transcript 1; qPCR, quantitative PCR; RPL13A, ribosomal protein L13A; RT-qPCR, quantitative RT-PCR.
The NEAT1 ΔPAS GBM cell lines created in this study harbor diminished NEAT1_1 with simultaneous increased NEAT1_2 levels, which elicited broad influences on the transcriptome. Similar numbers of DEGs were upregulated or downregulated and were enriched in distinct pathways (Fig. 6). The downregulated DEGs were involved in RNA processing, glial cell proliferation, and cell cycle modulation. Given the fact that NEAT1 overexpression is sufficient to promote cell growth, colony formation, as well as invasive migratory ability in a tumor type–specific manner (84, 85), diminished NEAT1_1 could contribute to part of these downregulated pathways (45, 86). In contrast, the upregulated DEGs were enriched in pathways involved in glial cell differentiation, gliogenesis, establishment of cell polarity, and regulation of cell migration. This raises an intriguing possibility that NEAT1 isoforms may impact distinct pathways in the cellular landscape of glioma. Given the multifaceted function of NEAT1 in gene regulation at the transcriptional and posttranscriptional levels (87, 88), molecular mechanisms underlying such glioma transcriptomic changes remain elusive. Whether and how changes in the aforementioned pathways caused by reciprocal alterations of NEAT1 isoforms contribute to glioma tumorigenesis will be the next challenge in future studies.
We next explored whether QKI-5 regulates the usage of the NEAT1 PAS through binding to the predicted nearby QREs. We searched the ENCODE Consortium dataset (Table S1) (67, 68) and found the highest QKI-5 UV-CLIP-seq peak near the 3′ end of NEAT1_1, which corresponds to a sequence region harboring three overlapping QREs immediately upstream of the NEAT1 PAS (Fig. 5A). This observation strongly suggests the direct interaction of QKI-5 with the NEAT1 primary transcripts at the QREs immediately adjacent to the NEAT1 PAS.
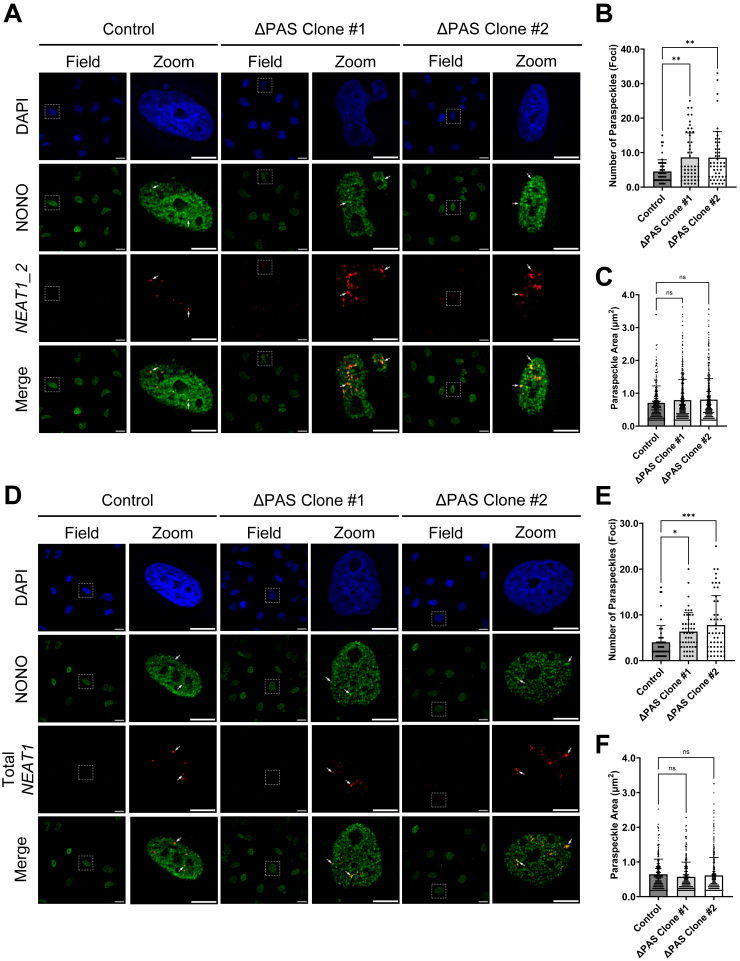
NEAT1_2 is the architectural lncRNA indispensable for paraspeckle formation, whereas NEAT1_1 is not essential for paraspeckle formation (27, 28, 29, 36, 43, 44). Paraspeckles are phase-separated nonmembranous nuclear bodies present only in a subpopulation of mammalian cells (29, 32, 35, 36, 37, 38, 43, 44, 58). Of note, no previous studies have examined paraspeckles in human glioma cells. We sought to characterize paraspeckle formation in U373 GBM cells based on the colocalized NEAT1_2 fluorescence in situ hybridization (FISH) signal with the immunofluorescence (IF) of NONO, an essential RBP component of paraspeckles (35, 38, 59). As shown in Figure 3A, FISH signals of a NEAT1_2-specific probe largely colocalizes with NONO IF in U373 GBM cells, clearly indicating detection of paraspeckle foci (Fig. 3, representative paraspeckle foci are indicated by arrows). Importantly, both NEAT1 ΔPAS clones show marked increases of colocalized NEAT1_2 RNA-FISH signals with NONO IF compared to the parent control (Fig. 3A). Quantification of confocal images shows increased paraspeckle numbers (NEAT1 ΔPAS clone #1: 8.620 ± 1.000 and NEAT1 ΔPAS clone #2: 8.531 ± 1.083) compared to control (4.520 ± 0.479) (Fig. 3B). However, the area of each paraspeckle foci in confocal images is not significantly changed in either NEAT1 ΔPAS clone (0.792 ± 0.031 μm2 and 0.807 ± 0.031 μm2) compared to control (0.709 ± 0.031 μm2) (Fig. 3C). Furthermore, no change in NONO protein levels is observed by immunoblot, indicating that increased paraspeckle formation in NEAT1 ΔPAS clones is not due to increased expression of NONO protein (Fig. S3).
In a single tube, 2 μg of isolated RNA was mixed with RNase inhibitor (Promega, N2615), DNase I (Invitrogen, 18047-019), 5× first strand buffer (Invitrogen, Y02321), and nuclease-free water, and incubated for 1 h at 37 °C. A 4:1 ratio of nuclease-free water and 1:1 ratio of phenol:chloroform (Invitrogen, 15593-031) was added to the tube and well mixed. Samples were centrifuged at 13,000 rpm for 5 min at 4 °C. The RNA was precipitated in a 10:1 ratio of 3M NaOAc and 3:1 ratio of 100% ethanol overnight at −80 °C. The following day, the samples were centrifuged at 13,000 rpm for 30 min at 4 °C. The RNA pellet was washed in 80% ethanol and centrifuged at 13,000 rpm for 5 min at 4 °C. The resulting RNA pellet was dissolved in nuclease-free water, and the quality was confirmed by agarose gel electrophoresis.
This section collects any data citations, data availability statements, or supplementary materials included in this article.
To determine the long-term effects on NEAT1 isoform imbalance caused by deletion of the NEAT1 PAS, we isolated and propagated two U373 NEAT1 ΔPAS clones based on PCR genotyping of the genomic DNA (Fig. S2A). Sanger sequencing of the PCR product confirmed the successful deletion of the NEAT1 PAS (Fig. S2B). RT-qPCR analysis clearly demonstrates diminished NEAT1_1 in both NEAT1 ΔPAS clones (Fig. 2, B and C), accompanied by a reciprocal increase of NEAT1_2 (Fig. 2, D and E). We also detected a significant increase in the levels of Total NEAT1 in the two NEAT1 ΔPAS clones compared to control (Fig. 2, F and G), possibly due to compensatory responses. These data suggest that usage of the NEAT1 PAS is a crucial mechanism that reciprocally controls the balance of NEAT1_1 and NEAT1_2 in GBM cells.
Protein lysates were boiled in reducing buffer and separated on 4 to 15% Mini-PROTEAN TGX polyacrylamide gels (Bio-Rad, 4568085) and then transferred to 0.45 μm polyvinylidene fluoride membranes (GE Healthcare Life Sciences). Membranes were incubated for 1 h in blocking buffer containing 10% nonfat milk in 0.1% PBS with tween. Membranes were incubated overnight at 4 °C in primary antibody diluted in blocking buffer. Primary antibodies were then detected with horse radish peroxidase–conjugated secondary antibodies and subjected to chemiluminescence detection with a Chem-iDoc Image System (Bio-Rad). The primary antibodies and dilutions used for immunoblotting are as follows: β-actin (mouse monoclonal, Sigma A5441, 1:10000), eIF5 (mouse monoclonal, Santa Cruz sc-28309; 1:10000), NONO (mouse monoclonal; Santa Cruz sc-166702; 1:1000), and QKI-5 (rabbit polyclonal, Bethyl A300-183A; 1:5000).
Model schematic for QKI-5 regulation of NEAT1 isoform biogenesis. Schematic depicting how QKI-5 binds to a QRE upstream of the NEAT1 PAS to promote the biogenesis of NEAT1_1 but loss of QKI-5 leads to increased NEAT1_2 formation. NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site; QKI, quaking; QRE, QKI recognition element.
To elucidate biological pathways affected by the deletion of the NEAT1 PAS in glioma, the PANTHER Gene Ontology (GO) program was utilized to identify molecular pathways impacted by the transcriptomic changes resulting from deletion of the NEAT1 PAS. Interestingly, most of the top hit pathways enriched for downregulated DEGs are implicated in ncRNA processing and RNA modification (Fig. 6E). Additional pathways downregulated include cell cycle check point and glial cell proliferation. In contrast, the upregulated DEGs are enriched in pathways implicated in cell polarity, matrix adhesion, glial cell differentiation, gliogenesis, and regulation of cell migration (Fig. 6F). We performed RT-qPCR and validated the RNA-seq identified upregulated DEGs including CD63 molecule (CD63), CD9 molecule (CD9), cadherin11 (CDH11), and glial fibrillary acidic protein (GFAP) (Fig. 6G), as well as downregulated DEGs represented by cell division cycle 20 (CDC20), dyskerin pseudouridine synthase 1 (DKC1), neugrin, neurite outgrowth associated (NGRN), and ribosomal protein L35A (RPL35A) (Fig. 6H). To our knowledge, this is the first study to characterize the functional impact of NEAT1 on the GBM transcriptome and identify biological pathways affected by increased NEAT1_2 accompanied by diminished NEAT1_1, which provides intriguing clues regarding the molecular mechanisms governed by NEAT1 isoforms in glioma tumorigenesis.
Primary GBM neurosphere cultures were raised from isolated surgical specimens donated for research with informed consent from patients and were collected and used according to recognized ethical guidelines in a protocol (IRB00045732) approved by the Institutional Review Board at Emory University. GBM cultures and normal human neural progenitor cells (Lonza) were maintained as per published protocols (57). The U373 human glioblastoma cells were propagated in Dulbecco's modified Eagle's medium/F12 (Corning, 10-013-CV) and supplemented with 10% fetal bovine serum (FBS) (GenClone 25-550). The LN229 and A172 human glioblastoma cell lines were propagated in Dulbecco's modified Eagle's medium/F12 (Corning, 10-013-CV) and supplemented with 10%FBS, 100 units/ml penicillin G, and 100 μg/ml streptomycin. For deletion of the NEAT1 PAS (NEAT1 ΔPAS), U373 cells that harbor Cas9 expression were transfected with two sgRNAs (Fig. S1A). For elimination of QKI-5 (ΔQKI5), U373 cells were transfected with two sgRNAs targeting the QKI exon 7c (Fig. S2A). For acute effects in bulk transfected cells, U373 cells were harvested 48 h after transfection for molecular analysis. Moreover, genetically edited clones harboring NEAT1 ΔPAS and ΔQKI-5 were isolated, respectively, and propagated for functional studies. For acute KD of QKI-5, a short siRNA (Table S3) specifically targeting QKI-5, or the Silencer Negative Control #1 (Invitrogen, 4390843) were transfected into U373, LN229, and A172 cells for two rounds on consecutive days. Cells were harvested 24 h after the second transfection for RNA analysis. A final concentration of 0.2 nM was used for all siRNA transfections. Sequences for the NEAT1 ΔPAS and ΔQKI-5 sgRNAs as well as the QKI-5 siRNA are shown in Table S3.
P. M. Z. and Y. F. conceptualization; Y. L. and B. Y. data curation; P. M. Z., L. K., Y. L., B. Y., G. J. B., R. D. R., and Y. F. formal analysis, P. M. Z., B. Y., R. D. R., and Y. F. funding acquisition; P. M. Z., L. K., G. Z., L. S., Y. L., R. D. R., and Y. F. investigation; P. M. Z., Y. L., L. S., B. Y., G. J. B., R. D. R., and Y. F. methodology; Y. F. project administration; P. M. Z., B. Y., G. J. B., R. D. R., and Y. F. resources; Y. F. supervision; P. M. Z., validation; P. M. Z., B. Y., G. J. B., R. D. R., and Y. F. visualization; P. M. Z. and Y. F. writing–original draft; P. M. Z., L. S., B. Y., G. J. B., R. D. R., and Y. F. writing–review and editing.
Many RBPs can regulate the recognition and usage of PAS in RNA 3′ end formation (44, 60, 61). Thus, we questioned whether and how GBM RBPs may regulate NEAT1 isoform balance through recognition and utilization of the NEAT1 PAS. We identified QKI recognition element (QRE) sequences located upstream of the human NEAT1 PAS (Fig. 4A), which are consensus RNA motifs containing ACUAAY-(1–20 nt)-UAAY for the binding of the glial RBP QKI encoded by a glioma risk gene (62, 63, 64, 65, 66). Alternative splicing of the QKI 3′ coding exons produces three QKI protein isoforms termed QKI-5, QKI-6, and QKI-7 (Fig. 4B) (65, 66). QKI-5 is nuclear localized while QKI-6 and QKI-7 are predominantly cytoplasmic (65, 66). To explore the potential roles of QKI in regulating NEAT1 isoforms, we initially performed siRNA KD specifically targeting QKI-5 mRNA (Fig. S4A), which led to a significant reduction of NEAT1_1 in multiple GBM cell lines (Fig. S4B). Conversely, NEAT1_2 levels and the ratio of NEAT1_2/NEAT1_1 are significantly increased (Fig. S4, C and D).
The lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) has been extensively studied and reported to be dysregulated in a variety of brain disorders and cancers. Human NEAT1 is located in chromosome 11 and transcribed by RNA polymerase II. Two distinct isoforms, NEAT1_1 and NEAT1_2, are generated due to alternative 3′ end processing (26, 27). NEAT1_1, a 3.7 kb transcript, is formed by conventional cleavage at the proximal polyadenylation site (PAS), followed by polyadenylation (26, 28). NEAT1_2, a 22.7 kb transcript, is formed by inhibiting the recognition and usage of the PAS, at the cost of decreased NEAT1_1 formation (27, 29). In contrast to polyadenylation, NEAT1_2 forms a tRNA-like structure at the 3′ end, which is cleaved by RNase P, and subsequently stabilized by the formation of a triple helix structure (30, 31). The roles of NEAT1 isoforms in nuclear organization are well characterized (32, 33, 34). NEAT1_2 is an architectural lncRNA essential for the formation of nuclear paraspeckles through the recruitment and organization of several RNA-binding proteins (35, 36, 37, 38, 39), which are found in a subpopulation of mammalian cells and implicated in cellular stress responses, cell differentiation, and cancer progression (40, 41, 42). Conversely, NEAT1_1, although present in paraspeckles, is not required for paraspeckle formation (27, 28, 29, 43, 44).
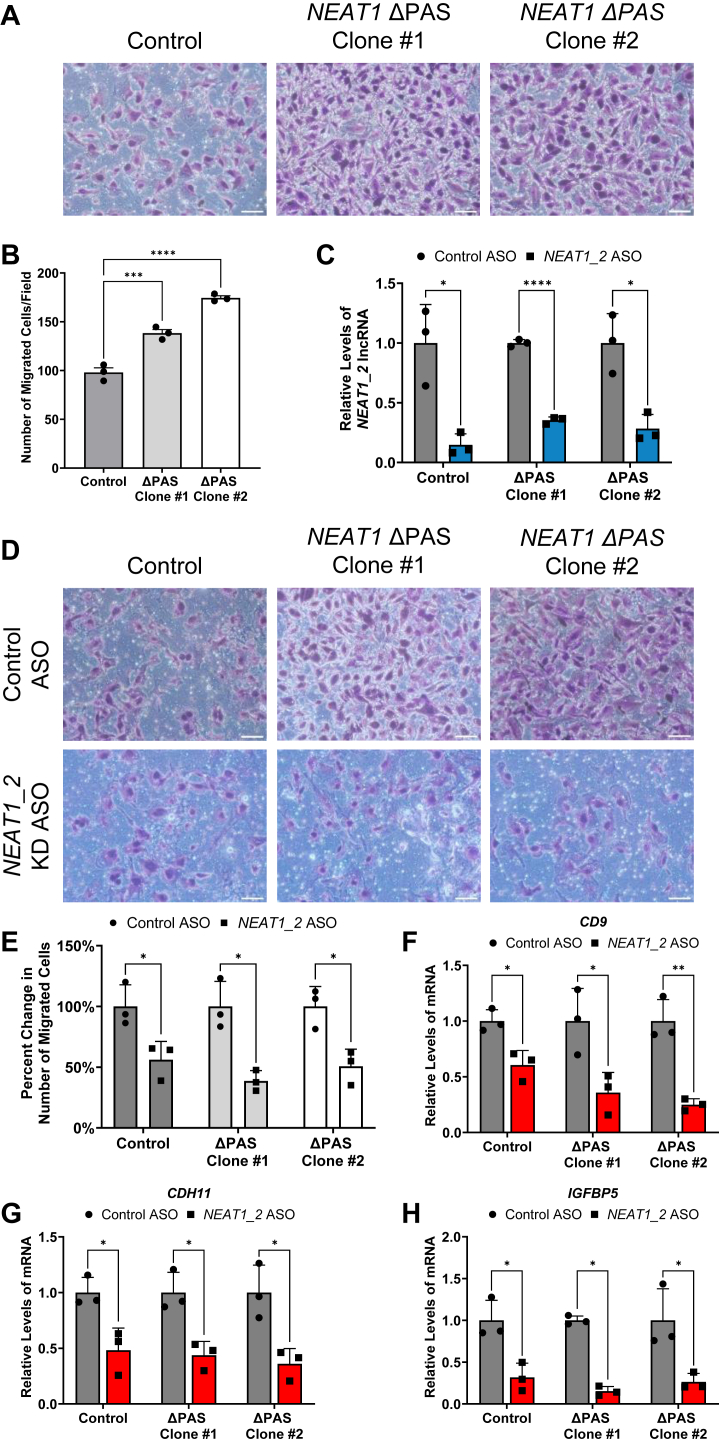
One of the top GO terms affected by NEAT1 ΔPAS is regulation of cell migration (Fig. 6F). We thus questioned whether and how glioma cell migration is altered by deletion of the NEAT1 PAS. A broadly used transwell assay was employed to evaluate cell migration. As shown in Figure 7A, increased cell migration is visible in both NEAT1 ΔPAS clones. Quantification of cell counts revealed a significant increase in the number of migrated cells in the two NEAT1 ΔPAS clones compared to the U373 control (Fig. 7B). To further determine whether the increased cell migration is due to the increase of NEAT1_2 upon NEAT1 ΔPAS, we utilized an ASO specifically targeting NEAT1_2. As shown in Figure 7C, the steady state levels of NEAT1_2 are significantly knocked down in the U373 control and two NEAT1 ΔPAS clones when treated with the NEAT1_2 ASO compared to a control ASO that contains a scrambled sequence. The transwell migration assay was conducted using the ASO-treated U373 control and NEAT1 ΔPAS clones. As shown in Figure 7D, a visible attenuation of migration is observed by the NEAT1_2 ASO in the U373 control and NEAT1 ΔPAS clones compared to treatment with the control ASO, which is confirmed by a statistically significant reduction in the number of migrated cells (Fig. 7E). Furthermore, we analyzed several NEAT1 ΔPAS upregulated DEGs enriched in the regulation of cell migration pathway. RT-qPCR revealed a significant decrease in the levels of CD9, CDH11, and insulin like growth factor binding protein 5 (IGFBP5) by the NEAT1_2 ASO in the U373 control and NEAT1 ΔPAS clones when compared to control ASO treatment (Fig. 7, F–H). Together these data demonstrate the increase of NEAT1_2 upon deletion of the NEAT1 PAS, not the loss of NEAT1_1, is responsible for promoting cell migration, which can be reversed by NEAT1_2 KD, suggesting that NEAT1_2 may play crucial roles in metastasis of GBM cells.
The high-throughput sequencing data, including RNA-seq, have are available at the Gene Expression Omnibus under the accession number GSE262598.
Cell migration assays were conducted using 8.0 μm Transwell inserts (Corning, 353097). Briefly, 1 × 105 cells in 300 μl serum-free media were plated in the upper chamber and 500 μl of 10% FBS-containing media was added to the lower chamber of a 24-well plate. Cells were incubated at 37 °C with 5% CO2 for 24 h after which the remaining cells in the upper chamber were removed with a cotton swab and the cells that migrated through the bottom of the membrane were fixed with 4% paraformaldehyde for 30 min. The cells were then stained with 0.1% crystal violet for 20 min and imaged with a microscope (Zeiss) at 20×. Three different fields were selected to count and measure the mean number of migrated cells using Image J software (https://imagej.net/software/fiji/). Three independent experiments were conducted, each in triplicate.
NEAT1 was reported to regulate splicing of specific RNAs through its bound RBPs (69, 70). To further investigate whether and how deletion of the NEAT1 PAS may alter transcriptome-wide mRNA splicing in glioma, we utilized the iRNA-seq package to analyze how splicing efficiency of the identified DEGs are affected (Fig. 6D). In the commonly upregulated DEGs identified in the NEAT1 ΔPAS clones, approximately 10% of DEGs show increased splicing efficiency, while 2% exhibit decreased splicing efficiency compared to control. Conversely, analysis of the commonly downregulated DEGs revealed approximately 9% of the DEGs show decreased splicing efficiency, with 2% showing increased splicing efficiency. These data suggest that altered splicing contributes to ∼10% of the transcriptomic alterations caused by deletion of the NEAT1 PAS.
Previous studies have reported an increase in NEAT1 levels in high-grade glioma specimens compared to control and low-grade glioma (47, 55, 72, 73, 74). However, these studies did not directly measure NEAT1_1. Hence, whether and how NEAT1 isoform balance is affected in glioma remained elusive. Taking advantage of the distinct 3′ end features of NEAT1_1 and NEAT1_2, our isoform-specific quantification assay reveals differential dysregulation of NEAT1 isoforms in GBM (Fig. 1). This observation raises an intriguing question as to whether the differential dysregulation of NEAT1 isoforms may contribute to the progression and severity of glioma. In addition, whether NEAT1 isoforms are also differentially dysregulated in other types of cancers and brain diseases warrants rigorous reinvestigation.
Efficient recognition and utilization of the proximal PAS is a crucial mechanism that governs NEAT1 isoform production. We demonstrate that CRISPR-Cas9–mediated genomic deletion of the NEAT1 PAS diminishes NEAT1_1 levels, which is accompanied by a significant increase in NEAT1_2 in GBM cells (Fig. 2), similar to previous studies which deleted the NEAT1 PAS in the human myelogenous leukemia haploid cell line HAP1 (29). Interestingly, deletion of the surrounding regions of the NEAT1 PAS led to opposite changes in NEAT1 isoform levels compared to that caused by deletion of the NEAT1 PAS alone (29). This observation suggests that RBPs bind to these regions to modulate the efficiency of the NEAT1 PAS usage which in turn governs NEAT1 isoform balance. One such RBP is HNRNPK, which binds to a sequence common in both NEAT1_1 and NEAT1_2, and competitively inactivates and arrests the NUDT21–CPSF6 complex (44). Consequently, 3′ end processing of NEAT1_1 at the proximal PAS is inhibited, leading to enhanced NEAT1_2 production in HeLa cells (44).
We utilized an oligo(dT)20 primer to reverse transcribe total RNA, by which complementary DNAs will be produced only from the RNAs that harbor a poly(A) tail, followed by qPCR with a primer set that amplifies the NEAT1 5′ region (primer set A, Fig. 1A Right) or a 3′ primer set specific for NEAT1_2 (primer set B). As shown in Figure 1B, NEAT1_2 is not detected in the human GBM cell lines U373, LN229, and A172, indicating that primer set A specifically detects NEAT1_1 in oligo(dT)20-mediated RT-qPCR. In a parallel experiment, random primers were used in RT followed by qPCR. Unlike in the oligo(dT)20-mediated RT-qPCR, NEAT1_2-specific primer pair B generates abundant RT-qPCR reads in the above GBM cell lines (Fig. 1C). Additionally, primer pair A detects both NEAT1 isoforms (Total NEAT1) in random primer-mediated RT-qPCR. The ribosomal protein L13A (RPL13A) mRNA carries a poly(A) tail, thus was detected in both the oligo(dT)20-mediated and random primer-mediated RT-qPCR reactions, serving as an internal reference for quantification of both NEAT1 isoforms.
For the quantification of lncRNAs and mRNAs, TRIzol-isolated and DNase-treated total RNA was reverse transcribed using either random primers (Promega, C1181) or oligo(dT)20 primers (Invitrogen, 18418020) with SSII reverse transcriptase (Invitrogen, 18064014) following the manufacturer’s instructions. qPCR was conducted with the Quantinova SYBR Green PCR Kit (Qiagen, 208056) using a CFX96 Real Time PCR System (Bio-Rad). RNA expression levels were normalized to either β-actin or ribosomal protein L13A, calculated by 2-ΔΔCT method. Primers used for RT-qPCR are summarized in Table S3.
In this study, we provide the first evidence that NEAT1 isoforms are differentially dysregulated in patient-derived GBM GSCs. Moreover, we demonstrate that recognition and usage of the NEAT1 PAS is a crucial mechanism that regulates NEAT1 isoform biogenesis in glioma under control of the GBM risk factor QKI-5. A working model for the mechanism of QKI-5 in the regulation of NEAT1 isoforms is illustrated in Figure 8. Finally, we identified broad changes in the GBM transcriptome and biological pathways in glioma tumorigenesis that are caused by forced biogenesis of NEAT1_2 with reciprocal reduction of NEAT1_1 and provide the first evidence that NEAT1_2 is responsible for driving GBM cell migration.
To investigate the functional importance of NEAT1 PAS utilization in the production of NEAT1 isoforms in glioma, we utilized CRISPR-Cas9 to delete the NEAT1 PAS (NEAT1 ΔPAS) in the U373 GBM cell line. To minimize off-target effects, two independent synthetic guide RNAs (sgRNAs) flanking the NEAT1 PAS were transiently transfected into cells that harbor Cas9 expression to delete the NEAT1 PAS (Figs. 2A and S1A). Forty-eight hours after transfection, RNA was extracted from the heterologous cell populations and subjected to the NEAT1 isoform–specific RT-qPCR assay. A significant decrease in steady state levels of NEAT1_1 is detected compared to control, accompanied with increased NEAT1_2 levels (Fig. S1, B and C). Consequently, the ratio of NEAT1_2/NEAT1_1 is significantly increased (Fig. S1D).
Mutations of putative QREs alter NEAT1 isoform biogenesis.A, eCLIP-seq reads at the NEAT1 gene. Largest peak maps to three QREs identified upstream of the NEAT1 PAS; sequences of the QREs provided below the indicated peak. B, schematic of the NEAT1 PAS reporter plasmid and RT-qPCR primers. Green arrows indicate the EGFP primer set used as an internal reference during RT-qPCR. Red arrows represent the primer set used to analyze cleavage activity at the NEAT1 PAS. Blue arrows indicate primer set used to measure NEAT1_2 levels from the reporter. Red letters in sequences below schematic indicate nucleotides subjected to site-directed mutagenesis to create the mutant plasmid construct, with QREs signified by underlined sections. C and D, RT-qPCR analysis of (C) the NEAT1 reporter transcript not cleaved at the PAS and (D) the reporter transcript containing the NEAT1_2 sequence downstream of the PAS. Results are derived from the mutant that lost the QREs compared to that of WT. Data are shown as mean ± SD from 4 biological replicates, normalized to the internal reference EGFP, and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗p < 0.05, ∗∗p < 0.01. NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site; RT-qPCR, quantitative RT-PCR; QRE, QKI recognition element.
Previous studies have specifically explored the role of NEAT1 in glioma migration. Zhou et al. observed decreased migration after treatment with a NEAT1 siRNA (55). However, the siRNA used targeted the common region of NEAT1_1 and NEAT1_2, therefore reducing both isoforms. We found that diminished NEAT1_1 and increased NEAT1_2 due to the loss of the NEAT1 PAS markedly enhanced migration of GBM cells, which was reversed by ASO KD of NEAT1_2 alone (Fig. 7). This result clearly demonstrated elevated NEAT1_2 is necessary and sufficient for driving GBM cell migration, regardless of diminished NEAT1_1. The fact that the regulation of cell migration GO pathway is enriched of upregulated DEGs in NEAT1 ΔPAS GBM cells and the reversal of DEGs indicated in cell migration by ASO KD of NEAT1_2 provides a novel molecular mechanism that further supports the oncogenic roles of NEAT1_2 in driving glioma cell migration and likely metastasis. These observations provide an intriguing mechanism for further understanding the distinct functions NEAT1 isoforms play in various aspects of glioma tumorigenesis.
We also conducted RNA-FISH using a probe targeting the overlapping region of NEAT1_1 and NEAT1_2 (Total NEAT1 FISH, Fig. 3D). Consistent with the increase in Total NEAT1 (Fig. 2), quantification of NONO IF colocalized with Total NEAT1 RNA-FISH revealed increased paraspeckle foci numbers in both NEAT1 ΔPAS clones (NEAT1 ΔPAS clone #1: 6.340 ± 0.583 and NEAT1 ΔPAS clone #2: 7.760 ± 0.910) compared to control (4.000 ± 0.518) (Fig. 3E). Again, the area of each paraspeckle is not significantly changed in either U373 NEAT1 ΔPAS clone (0.570 ± 0.024 μm2 and 0.613 ± 0.026 μm2) compared to control (0.645 ± 0.030 μm2) (Fig. 3F). Together, these data demonstrate that the increase of NEAT1_2 in response to deletion of the NEAT1 PAS is sufficient to enhance paraspeckle formation in human GBM cells despite the decrease in levels of NEAT1_1.
If you have not received this email or if you are having problems logging in, reset your password using the link below or contact us.
The roles of RBPs involved in glioma tumorigenesis in regulating NEAT1 isoform biogenesis through interacting and modulating the usage of the NEAT1 PAS has not been studied. The glioma risk factor RBP QKI drew our attention because multiple consensus QREs are found immediately upstream of the human NEAT1 PAS. Interestingly, the mouse Neat1 PAS region does not harbor these predicted QREs. Among the three QKI protein isoforms, nuclear QKI-5 is the predominant isoform expressed in the U373 GBM cells. Moreover, the strongest QKI-5 UV-CLIP-seq peak was mapped to the QREs near the NEAT1 PAS (Fig. 5). Indeed, elimination of QKI-5, by either transient siRNA KD of QKI-5 in multiple GBM cell lines (Fig. S4) or CRISPR-Cas9 deletion of the QKI-5–specific exon 7c in the U373 GBM cell line (Fig. 4) significantly reduces endogenous NEAT1_1 but reciprocally elevates NEAT1_2. Moreover, mutagenesis of the QREs clearly demonstrates the suppression of cleavage at the NEAT1 PAS in a reporter transcript (Fig. 5). Thus, despite the presence of smaller QKI-5-UV-CLIP-seq peaks that may involve QKI in NEAT1 stability, the QRE-dependent interaction of QKI-5 near the NEAT1 PAS which facilitates 3′ end processing of the reporter clearly demonstrates critical roles of QKI-5 in modulating NEAT1 PAS recognition and usage. To our knowledge, this is the first example of QKI-5 in regulating the biogenesis of glioma-associated lncRNAs, despite the well-characterized roles of QKI-5 in regulating numerous mRNAs and circRNAs (75, 76). In this regard, the frequent deletion of the QKI locus found in GBMs (62, 63) may affect NEAT1 isoform biogenesis and glioma tumor development.
RNA splicing efficiency was determined using the package iRNA-seq. Briefly, all significant DEGs (FDR < 0.05) in the two NEAT1 ΔPAS clones were analyzed for exonic and intronic expression. The exonic expression represented spliced RNA while the intronic expression represented unspliced RNA. Splicing efficiency was calculated using the following formula:
Alterations of NEAT1 isoform levels influence paraspeckle abundance.A, representative RNA-FISH images of U373 GBM control and two NEAT1 ΔPAS clones stained for NEAT1_2 (red) and immunofluorescence of NONO (green). Nuclei were stained with DAPI (blue). Arrows indicate representative paraspeckles, identified by colocalization of the NEAT1 FISH probe and NONO IF. Field image scale bar represents 25 μm; zoomed image scale bar represents 10 μm. B and C, dot plot quantification of (B) number of paraspeckles (colocalized foci) and (C) area of individual paraspeckle (μm2) in control and two U373 NEAT1 ΔPAS clones. Data are shown as the mean ± SD of 50 nuclei. Statistical significance was calculated using one-way ANOVA with Dunnett multiple comparison’s test, ns = not significant, ∗∗p < 0.01. D, representative RNA FISH images of U373 GBM control and two NEAT1 ΔPAS clones stained for Total NEAT1 (red) and immunofluorescence of NONO (green). Nuclei were stained with DAPI (blue). Field image scale bar represents 25 μm; zoomed image scale bar represents 10 μm. E and F, dot plot quantification of (E) number of paraspeckles (colocalized foci) and (F) area of individual paraspeckle (μm2) in control and two U373 NEAT1 ΔPAS clones. Arrows indicate paraspeckles. Data are shown as the mean ± SD of 50 nuclei. Statistical significance was calculated using one-way ANOVA with Dunnett multiple comparison’s test, ns = not significant, ∗p < 0.05 and ∗∗∗p < 0.001. DAPI, 4′,6-diamidino-2-phenylindole; GBM, glioblastoma multiforme; IF, immunofluorescence; NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site.
While some studies reported tumor-related functions of NEAT1 isoforms (51, 52), most studies only characterized the dysregulation and function of Total NEAT1 (53, 54, 55). This limits our understanding of NEAT1 isoform function and the ability to develop diagnostic biomarkers and treatments based on isoform-specific roles and mechanisms. In human GBM, NEAT1 was reported to be aberrantly upregulated and thought to enhance glioma progression (56). However, whether NEAT1 isoforms are equally or differentially dysregulated has not been determined. In addition, whether NEAT1_1 and NEAT1_2 play distinct roles in tumorigenesis is not understood, due to the lack of specific functional analyses of each NEAT1 isoform. Furthermore, how NEAT1 dysregulation impacts the tumor transcriptome and functional pathways remains elusive.
Deletion of the NEAT1 PAS alters isoform steady state levels.A, schematic depicting the deletion of the NEAT1 PAS by CRISPR-Cas9. sgRNAs are depicted by brown and orange arrows and cleavage sites represented as scissors. RT-qPCR detection primers are depicted by green arrows. B and C, detection of NEAT1_1 steady state levels upon the deletion of the NEAT1 PAS. Data are shown as mean ± SD from 5 biological replicates, normalized to RPL13A and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗∗∗∗p < 0.0001. D and E, RT-qPCR analysis of NEAT1_2 steady state levels upon the deletion of the NEAT1 PAS. Data are shown as mean ± SD from 5 biological replicates, normalized to RPL13A and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗p < 0.05 and ∗∗∗p < 0.001. F and G, detection of Total NEAT1 steady state levels in two isolated U373 NEAT1 PAS deletion clones. Data are shown as mean ± SD from 6 (control) and 5 (ΔPAS) biological replicates, normalized to RPL13A, and compared using the ΔΔCT method. Unpaired Student’s t test was used, ∗p < 0.05 and ∗∗p < 0.01. NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site; RPL13A, ribosomal protein L13A; RT-qPCR, quantitative RT-PCR; sgRNA, synthetic guide RNA.
Official websites use .gov A .gov website belongs to an official government organization in the United States.
In this study, we specifically quantified each NEAT1 isoform and observed differential dysregulation of NEAT1 isoforms at steady state levels in patient-derived human GBM gliomasphere cultures (GBM GSCs). CRISPR-Cas9–mediated deletion of the NEAT1 PAS reduced NEAT1_1 and reciprocally increased NEAT1_2, resulting in paraspeckle hyperformation in GBM cells. Furthermore, we found that the RBP quaking (QKI), a glioma risk factor, regulates the biogenesis of NEAT1 isoforms through facilitating NEAT1 PAS usage in glioma cells. We characterized alterations of the glioma transcriptome and identified functional molecular pathways caused by altered NEAT1 isoforms. Finally, we showed that the increase of NEAT1_2 is responsible for enhanced glioma cell migration in culture, which is reversed by antisense oligonucleotides (ASOs) that specifically KD NEAT1_2. Together, our findings reveal new mechanisms that regulate NEAT1 isoform biogenesis and their functional impacts on the glioma transcriptome and migration.
Increased NEAT1_2 levels are responsible for promoting GBM cell migration.A, images of a transwell migration assay measuring migrated cells in the U373 control and the two NEAT1 ΔPAS clone cells. The scale bar represents 20 μm. B, quantification of the number of migrated cells per field of view taken in three independent replicates of the transwell migration assay for the U373 control and NEAT1 ΔPAS clones. One-way ANOVA with Dunnett multiple comparison’s test was used, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001. C, RT-qPCR analysis of NEAT1_2 steady state levels upon transfection of the control ASO or NEAT1_2 ASO in the U373 control and NEAT1 ΔPAS clones. Data are shown as mean ± SD from 3 biological replicates, normalized to RPL13A, and compared using the ΔΔCT method. Unpaired Student’s t test with Holm-Šídák multiple comparison’s test was used, ∗p < 0.05 and ∗∗∗∗p < 0.0001. D, images of transwell migration assay measuring the cell migration in the control ASO or NEAT1_2 ASO-treated U373 parent control and NEAT1 ΔPAS clone cells. The scale bar represents 20 μm. E, quantification of the number of migrated cells per field of view taken in three independent replicates of the ASO-treated transwell migration assay. Unpaired Student’s t test with Holm-Šídák multiple comparison’s test was used, ∗p < 0.05. F–H, RT-qPCR analysis of (F) CD9, (G) CDH11, and (H) IGFBP5, DEGs enriched in the regulation of cell migration pathway. Data are shown as mean ± SD from 3 biological replicates, normalized to RPL13A, and compared using the ΔΔCT method. Unpaired Student’s t test with Holm-Šídák multiple comparison’s test was used, ∗p < 0.05, ∗∗p < 0.01. ASO, antisense oligonucleotide; DEG, differentially expressed gene; GBM, glioblastoma multiforme; IGFBP5, insulin like growth factor binding protein 5; NEAT1, nuclear paraspeckle assembly transcript 1; PAS, proximal polyadenylation site; RPL13A, ribosomal protein L13A; RT-qPCR, quantitative RT-PCR.
To directly test whether these QREs can regulate NEAT1 PAS usage by QKI-5, we engineered a NEAT1 PAS reporter construct (Fig. 5B). A 941 bp DNA fragment containing sequences flanking the NEAT1 PAS, including all three predicted QREs, was PCR amplified and inserted downstream of EGFP (Fig. S6A). Additionally, as shown in Figure 5B, a mutant reporter was created in which all three QREs were mutated, as confirmed by sequencing of the construct (Fig. S6B). In both the WT and mutant constructs, the SV40 PAS was included downstream of the NEAT1 fragment.
This work was supported by National Institutes of Health under the following awards: National Institute of Neurological Disorders and Stroke (R01NS110110 to Y. F., R01NS118819 to Y. F and B. Y., and R01NS100967 to R. D. R.), National Institute on Aging (R01AG078937 to B. Y.), and National Institute of Mental Health (F31MH127915 to P. M. Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
To determine the long-term effects of QKI-5 loss on NEAT1 isoforms, we utilized CRISPR-Cas9 to delete exon 7c which is specific for QKI-5 (ΔQKI-5) (Figs. 4B and S5A). A ΔQKI-5 U373 clone was isolated and propagated, in which deletion of exon 7c was evident, based on the reduced RNA-seq reads specifically mapped to exon 7c, with no change in exons 7a or 7b in the other QKI isoforms (Fig. S5B). Furthermore, RT-qPCR analysis demonstrated the loss of QKI-5 mRNA (Fig. 4C), and the loss of QKI-5 protein was validated by immunoblot analysis (Fig. 4D). As a consequence, a significant decrease in NEAT1_1 is observed in the ΔQKI-5 clone compared to control (Fig. 4E). Conversely, NEAT1_2 levels as well as the ratio of NEAT1_2 to NEAT1_1 are significantly increased in the ΔQKI-5 clone (Fig. 4, F and G). These data suggest QKI-5 promotes the biogenesis of NEAT1_1 and reciprocally suppresses the production of NEAT1_2, most likely through enhancing NEAT1 PAS usage.
Using the above assay, we found that both NEAT1_1 and NEAT1_2 are significantly increased at steady state levels in human GBM GSCs derived from surgically resected tumor tissue from six different patients (57) compared to the healthy human neural progenitor cell control (Fig. 1, D and E). However, the fold increase of NEAT1_1 exceeds that of NEAT1_2. As a result, the ratio of NEAT1_2 to NEAT1_1 in each GSC line is significantly reduced as compared to the control (Fig. 1F). Together, these data established a method that distinctly quantifies each NEAT1 isoform, which reveals imbalanced dysregulation of NEAT1 isoforms in patient-derived GBM GSCs.
Poly(A) RNA was isolated using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490L), following the manufacturer’s instructions.
Cultured cells were harvested and centrifuged at 5000 rpm. The resulting cell pellets were resuspended in TRIzol (Invitrogen, 15596018) for 5 min. A 1:5 ratio of chloroform (Thermo Fisher Scientific, CX-1055-9) was added, mixed, and incubated for 15 min at room temperature. Samples were centrifuged at 13,000 rpm for 15 min at 4 °C. The aqueous layer was transferred to a clean tube to which a 1:1 ratio of isopropanol (Thermo Fisher Scientific, A-451-4) was added. The solution was incubated for 15 min at room temperature and then centrifuged at 13,000 rpm for 15 min at 4 °C. The resulting RNA pellet was then washed with 80% ethanol and centrifuged at 13,000 rpm for 5 min at 4 °C. The pellet was dissolved in nuclease-free water, quantified by BioDrop, and the quality verified by agarose gel electrophoresis or by Bioanalyzer.



 Neil
Neil 
 Neil
Neil